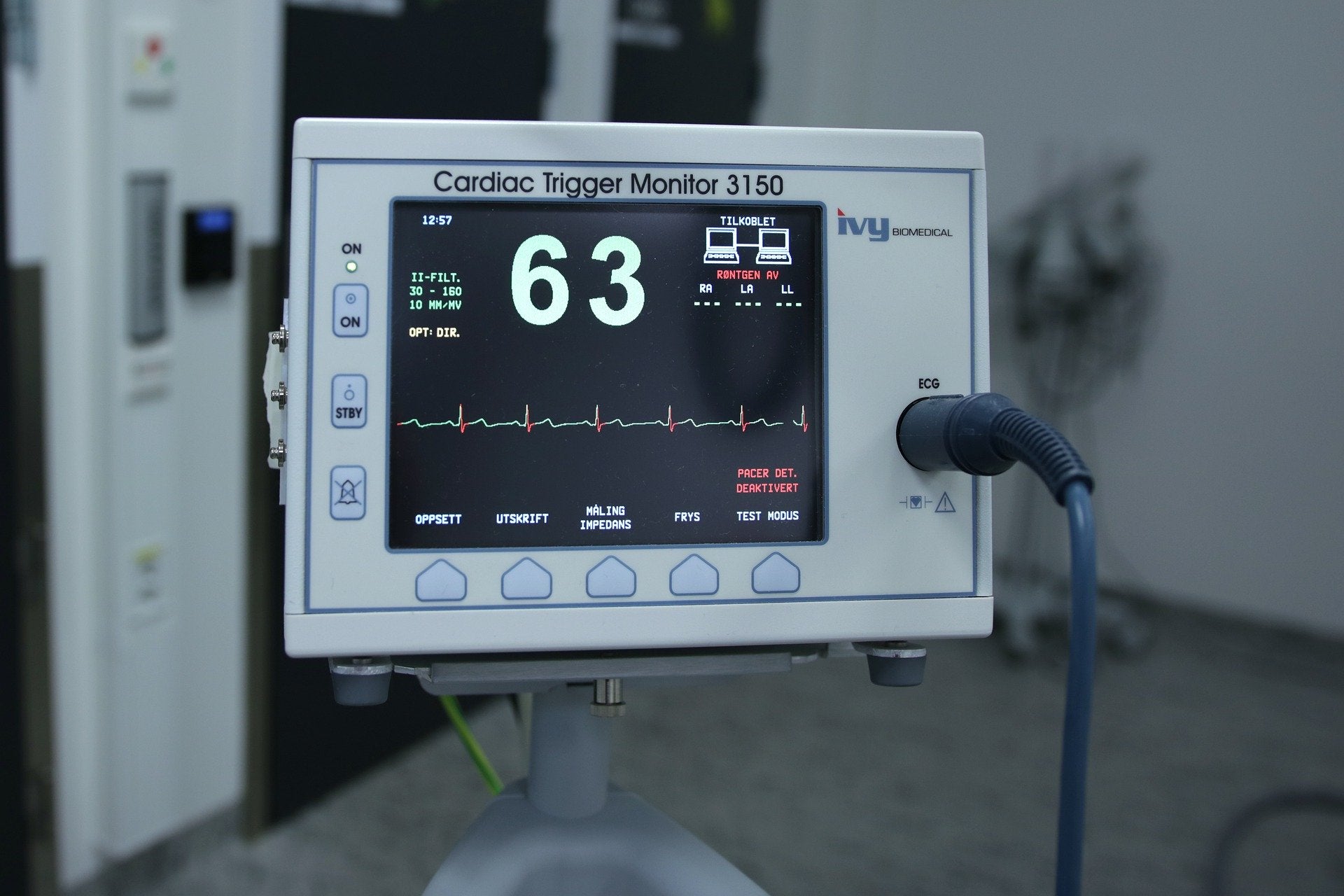
With the coronavirus crisis creating urgent need for ventilators and respiratory equipment across Europe, it brings 2017’s game-changing EU Medical Device Regulation into sharp focus as the manufacture of the machines commences in many disparate industries. Professor Herman Pieterse, of the University of Ghent and Profess Medical Consultancy, spoke to Medical Device Developments in 2018, outlining the considerable demands being made on device companies and how they were adapting to change.
Much has already been written about the EU Medical Device Regulation (MDR), which has now formally taken the place of the 1995 Medical Device Directive (MDD). Its purpose is to bring similar safety controls and transparency to medical device products as to those governing medicinal products, and in the process, make manufacturers and overseeing authorities more accountable for conducting all the right checks across a product’s life cycle.
Although the regulation has been active [since 25 May 2017], a realistic transition phase has been allowed. This is just as well, given the significant changes that are required. Under the regulation, which encompasses a whole raft of different standards, all MDD certificates will expire by May 2024, and final sales of stock will need to be made by June 2025. As of May 2017, notified bodies have been able to request redesignation, and manufacturers are able to start placing devices on the market under the new regulation. By 2020, this will be compulsory.
The reasons for introducing MDR are well known. While drug-oriented life sciences have come a long way in terms of enhanced regulatory controls and transparency over the past decade, the medical device industry still has a lot of catching up to do; and in Europe, MDR is the mechanism that will help ensure this happens. Serious safety incidents and patient deaths resulting from poorly designed devices slipping through the nets of monitoring bodies have acted as a stark reminder of the dangers of inadequately monitored product manufacture and supply – especially for devices embedded in the human body. The pivotal case was the 2010 Poly Implant Prostheses (PIP) breast implant scandal, involving devices fraudulently manufactured using unapproved silicone gel, which had dire consequences. Following the introduction of MDR, events like this should never be able to happen again.
EU Medical Device Regulation and a streamlining of notified bodies
Among the changes introduced with MDR is a planned streamlining of notified bodies. In Europe, there are 70–80 of these entities, each boasting a lot of experience, but not necessarily in specialist areas, such as knowledge of clinical evidence relating to breast implants. A significant rationalisation and recertification of these organisations will see numbers fall to roughly 20 – and the remaining organisations will need to demonstrate high levels of expertise and will be closely overseen to ensure they are consistently vigilant in their checks.

Class III medical devices in particular (such as surgical devices, implants and hip replacements) will be subject to much more stringent surveillance from now on. One of the uncomfortable discoveries in the wake of the PIP breast implant crisis was how much had slipped past the overseeing authorities because of a lack of rigour (like an absence of routine annual reviews of products on the market, and even adequate proof of efficacy before devices were accepted for marketing) and a lack of product traceability.
In the interests of unequivocal device traceability across entire supply chains and throughout their full life cycles, the EU has prescribed that any medical device made available in an EU member state must be accurately listed in a central European portal, the purpose-designed Eudamed database of medical devices.
Transparency will allow the alarm to be sounded at the first hint of a problem
Pivotal in enabling all of this will be a device’s unique device identifier (UDI) – with direct implications for the packaging and labelling of medical devices, whether they are relatively innocuous Class I items (like stethoscopes, crutches and hospital beds), or more safety-sensitive Class II ventilators and III pacemakers or contraceptive implants. As well as preventing manufacturers and overseeing bodies from sidestepping their responsibilities, the increased transparency will allow the alarm to be sounded at the first hint of a problem with a product or batch once it is out in the market, or even before. This will prevent the damage associated with faults, contamination or other potential issues, and give manufacturers more control over rapid recalls – reducing those companies’ exposure to risk.
EU Medical Device Regulation-based UDI requirements follow existing US UDI requirements
The good news is that MDR-based UDI requirements will closely follow existing US UDI requirements; there is no EU-specific deviation that would require manufacturers to formulate new processes for European markets. This is important in the interests of global harmonisation and consistent raising of quality standards. Certainly, it is not just European suppliers that are affected by MDR.
One of the knock-on effects of this increased vigilance and monitoring of devices sold across the EU is that, for example, US and Asian manufacturers and distributors will need authorised representatives to deal with the expected increase in liability issues when supplying to Europe. European manufacturers must also appoint designated responsible or qualified persons who will be accountable for regulatory compliance – as is the case in the pharmaceutical industry.
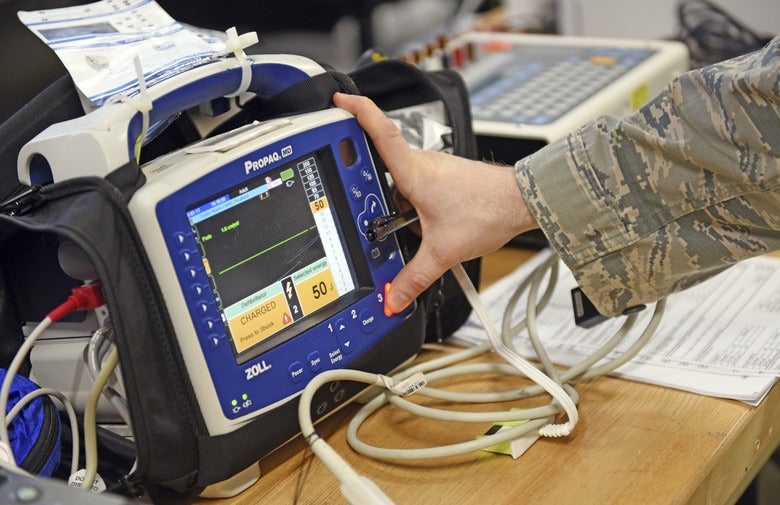
All of this places new, considerable demands on medical device companies. Having an effective, compliant UDI, for instance, requires that processes are set up to manage structured data so that the UDI can be assigned, recognised, tracked and reported on – from the assembly line to final use.
MDR requires that a UDI label be directly attached to a medical device or to its packaging and include two identifiers: a device identifier (DI), which identifies the labeller and the specific version or model of a device; and a production identifier, the variable portion of a UDI referring to factors such as the given lot or batch number, serial number, date of manufacture, expiry date and so on.
As devices are produced, the manufacturer will need to submit the UDI-DI and additional product data, such as a single registration number and whether the item contains latex or other specific substances, to the central Eudamed database, where it can be accessed by members of the public and healthcare professionals.
Compliance with MDR is a prerequisite if manufacturers are to continue to sell their products in a given market
Translations have long been an important element within the medical device industry, and nowhere more so than in Europe, where so many different languages are spoken. Under MDD, language requirements were the responsibility of each member state, but the new MDR requires that labelling is in the national language and “clearly comprehensible to the intended user or patient”.
Compliance with MDR is a prerequisite if manufacturers are to continue to sell their products in a given market. Unless they invest in the right resources and update their ways of working, they could lose EU market access – for existing products and those new to market. Estimates suggest 30% of current medical devices could disappear from the market if companies are not on top of the new requirements. Market access planning could be dictated by companies’ ability to find and appoint qualified persons, or access to notified bodies
Certainly, there are a great many considerations to absorb, and companies are advised not to wait until closer to 2020 before they adapt in earnest, given everything that is at stake.
It is important to remember why these updated measures have been brought in – to restore public trust and improve patient safety. So manufacturers that take early action have much to gain through being seen to take these priorities seriously.
This article originally appeared in the 2018 Volume 2 edition of Medical Device Developments. The full issue can be viewed here.




